Transient hypogammaglobulinemia of infancy
Abstract
Transient hypogammaglobulinemia of infancy (THI) was first described as a distinct entity by Gitlin and Janeway in 1956. Although THI has been recognized for many years, and despite significant progress in understanding the molecular basis and identifying the genes involved in the pathogenesis of many other forms of humoral immunodeficiencies, not much is known about this specific entity. This article summarizes the definition of THI, possible etiologies, clinical manifestations, treatment, and prognosis.
Antibody responses to specific antigens in neonates and infants
Serum IgG levels at birth are equal to or slightly higher than maternal serum IgG levels, which is the consequence of the transfer of maternal IgG across the placenta during the third trimester of pregnancy (Kohler and Farr 1966). Accordingly, premature infants have lower IgG concentrations. Hobbs and David (1967) demonstrated that premature infants at 30–32 weeks of gestation have cord IgG concentrations of only approximately 400 mg/dL.
The basic cellular elements of the immune system, including Ig-bearing B cells, are well established by 15 weeks of gestation. Nevertheless, serum immunoglobulin concentrations remain very low (below 100 mg/dL) until 18–20 weeks of gestation. Small for gestational age (SGA) neonates may have somewhat lower IgG levels than full-term neonates, reflecting possible impaired placental transport (Shapiro et al. 1981; Einhorn et al. 1987).
Full-term neonates can produce specific antibodies to T-cell dependent protein antigens soon after birth. In fact, even premature infants as young as 24 weeks of gestation and SGA infants appear to respond in a similar manner to protein vaccines (Smolen et al. 1983; Koblin et al. 1988). These and several other studies (Dancis et al. 1953; Bernbaum et al. 1985; McGeady 1987) suggested that the interval from birth to immunization is a more important factor than the gestational age itself. In contrast, the response to polysaccharide T-cell independent antigens appears later, at approximately 18–24 months of age (Smith et al. 1973; Hayakawa et al. 1981). The same pattern has been observed in premature and SGA infants. This could be partially explained by a preferential active transport of IgG1 and IgG3 across the placenta because of a higher affinity of the Fc receptors on trophoblasts for these IgG subclasses (McNabb et al. 1976; Einhorn et al. 1987; Van de Winkel and Anderson 1991) and the slow rise of IgG2 and IgG4 concentrations. IgG2, and to a lesser degree IgG4, are the predominant antibodies produced after natural exposure to polysaccharide antigens (Ochs and Wedgewood 1987).
After birth, the levels of maternally derived IgG decline rapidly and reach their lowest point of approximately 400 mg/dL at 3–6 months. At the same time, the infant's own production of IgG is not fully developed. This sequence of events is generally accepted as representing normal “physiologic” hypogammaglobulinemia. This phenomenon is more pronounced in premature infants, as they have proportionally lower IgG levels at birth and values reach an even lower nadir at 3 months of age. Ballow et al. (1986) reported concentrations of 60 mg/dL and 100 mg/dL at 3 months of age in premature infants born at 25–28 and 29–30 weeks of gestation, respectively. Interestingly, most of these infants did not have a higher incidence of infections, despite these very low IgG concentrations. By the age of 1 year, the total IgG concentration is approximately 60% of that of adults.
There is also a different pattern of progression in the synthesis of the Ig subclasses after birth. IgG1 and IgG3 reach adult concentrations earlier than IgG2 and IgG4, at 8 and 10–12 years of age, respectively (Ochs and Wedgewood 1987).
IgA, IgM, IgD, and IgE are not placentally transferred under normal circumstances. In fact, the presence of elevated levels of IgM or IgA in cord blood suggests intrauterine infection (Alford et al. 1975). In normal infants, the IgM concentration rises rapidly for the first month after birth, presumably in response to the massive antigenic stimulation of the new environment, and then more gradually up to 60% of the adult level by the age of 1 year. Serum IgA levels rise slower than IgM, up to values of only 20% of those of adults by 1 year of age, and then continues to rise progressively throughout adolescence (Wilson et al. 1996).
Definition
Transient hypogammaglobulinemia of infancy (THI) has been classically defined as an accentuation and prolongation of the “physiologic” hypogammaglobulinemia of infancy normally observed during the first 3–6 months of life (Gitlin and Janeway 1956). Nevertheless, the criteria for the diagnosis are not well standardized. In 1952, the World Health Organization (WHO; World Health Organization 1992) included both diminished IgG and IgA when defining the criteria, whereas the latest published report (IUS Scientific Committee 1999) stated that “one or more classes of Ig may be very low–within an immunodeficient range.” Most of the reports used low IgG levels as a mandatory inclusion criterion, whereas others have used other Ig isotypes such as low IgA (McGeady 1987). The fact that IgA levels reach only 20% of adult values at 1 year of age together with the high variability in the normal range of IgA (Wilson et al. 1996) levels make it an unreliable determinant for diagnosis of THI. Although most of the published studies (Tiller and Buckley 1978; McGeady 1987; Dressler et al. 1989; Dalal et al. 1998) require that the concentration of at least one class of Ig be more than 2 standard deviations (SD) below the mean for age-matched controls, others have used 3 SD for definition (Siegel et al. 1981). We suggest using a definition based on IgG levels that fall at least 2 SD below the mean for aged-matched controls as an invariant criteria with or without diminished values of other immunoglobulin isotypes.
Incidence
The precise frequency of THI remains undetermined. Tiller and Buckley (1987) reported that only 11 cases of THI were identified among more than 10 000 patients, whose sera were sent for Ig measurements, suggesting that THI is not a common entity. Dressler et al. (1989) supported this assumption by finding only 5 new cases in more than 8000 samples over a period of 11 years. Walker et al. (1994) reported 15 patients with proven THI and another 25 patients with possible THI out of 2468 referrals over a 10-year period. This shows an incidence of 23 or 61 per 106 live births, respectively (Walker et al. 1994). An overview of a nationwide survey on primary immunodeficiency in Japan indicated that 18.5% of patients were diagnosed as having THI (Hayakawa 1981).
From the 1632 infants and children who were referred to the immunology clinic at the Hospital for Sick Children, Toronto, Canada, between 1985 and 1995, 35 were diagnosed as having THI (Dalal et al. 1998). The disparity of THI incidence among various centers can be best explained by the lack of strict criteria for the diagnosis of this disorder.
Cohorts from the last few years demonstrate a disproportionate number of male infants, (up to 70%) (Van Winkle et al. 1994; Qian et al. 2009; Karaca et al. 2010; Keles et al. 2010). This might suggest that THI is more common in males, or it can be that the normal values we use for evaluation should reflect not only age but also sex.
Etiology
The cause of THI remains unknown despite numerous pathogenic mechanisms that have been proposed. Fudenberg and Fudenberg (1964) demonstrated that gamma globulin (Gm) antigenic determinants present on human fetal IgG molecules, but not in the mother, can induce the synthesis of maternal IgG anti-Gm antibodies during pregnancy. They concluded that such alloantibodies might cross the placenta and cause transient suppression of fetal immunoglobulin production. A prospective study on this issue, however, did not support this hypothesis (Nathenson et al. 1971). Another theory was suggested by Soothill (1968) after his finding of THI in the relatives of patients with a variety of immunodeficiency (ID) disorders. He suggested that THI is a manifestation of genetic heterozygosity for some other ID diseases. Although this remains a possibility, an example of such a trend has not been found to date. Siegel et al. (1981) proposed that a defect in helper-T-cell maturation could be the basis of THI. This conclusion was supported by a finding of normal B cell number and function together with numerical and functional deficiency in CD4+ T helper cells. Moreover, they demonstrated that this defect was transient, as patients who had recovered from THI had normalized CD4+ T-cell number and function. Unfortunately, this observation has not been supported by many other studies that followed (Rieger et al. 1977; Dressler et al. 1989; Kowalczyk et al. 1997; Dalal et al. 1998).
More recent work has suggested a role for cytokines in the pathogenesis of THI. Kowalczyk et al. (1997) demonstrated an enhanced production of tumor necrosis factor (TNF)-alpha, TNF-beta, and IL-10 in THI patients, whereas secretion of other cytokines such as IL-1, IL-4, and IL-6 was essentially similar to controls. They also demonstrated that exogenously added TNF-alpha and TNF-beta inhibited IgG and IgA secretion by pokeweed mitogen-stimulated mononuclear cells. Moreover, normalization of serum IgG levels in THI patients was associated with a decrease in TNF-alpha and TNF-beta production, but IL-10 production remained unchanged. They concluded that TNF may be involved in the regulation of IgG and IgA production and the balance between TNF production (suppressing IgG synthesis) and IL-10 (inducing IgG switch) may be important for the normal development of IgG-secreting B cells.
Recently, Rotwalczyk et al. (2011) reported a transient increase in absolute numbers of regulatory T-cells. The mechanism responsible for this finding is still unknown. Because TGFβ signaling is required for the generation of T regulatory cells it was speculated that mutations in the TGFβ gene may explain this finding. Unfortunately, mutation analysis of this gene did not support this assumption (Rutkowska et al. 2011, 2013).
Clinical manifestations
THI has been described in 2 distinct groups of infants based on the circumstances in which a diagnosis was obtained. The first group consisted of relatives of patients with other well-defined ID diseases. Most appeared to be in good health with no history of recurrent infection, and diagnosis was reached only because they were screened for Ig concentrations. These patients normalized their Ig levels and clinically remained asymptomatic (Soothill 1968; Tiller and Buckley 1978). The significance of this finding remains unknown. In contrast, the second group consisted of infants whose Ig measurement was determined because of recurrent infections starting early in life. The spectrum of severity and type of infections varied from otitis media and bronchitis to life-threatening invasive infections such as bacterial meningitis (Wilson et al. 1996). The initial indication for immunologic evaluation was recurrent upper respiratory infections, with or without ear or chest infections (Dalal et al. 1998). Other indications included severe Varicella (Adenyi-Jones et al. 1992), prolonged oral thrush (Adenyi-Jones et al. 1992), polio-like disease (Adderson et al. 1991), and invasive infection (bacteremia with cellulitis and meningitis) (Adenyi-Jones et al. 1992).
Atopic diseases such as bronchial asthma, allergic bronchitis, and atopic dermatitis were linked to hypogammaglobulinemia in several studies, with varying prevalence. In the latest reports 26.5%–63% of THI patients had atopic diseases (Tiller and Buckley 1978; Dalal et al. 1997; Kiliç et al. 2000; Doğu et al. 2004; Kidon et al. 2004; Papadopoulou et al. 2005; Whelan et al. 2006; Agondi et al. 2010; Keles et al. 2010). The underlying mechanism for this link is not yet understood.
Another manifestation observed infrequently is hematologic abnormalities. Transient neutropenia has been observed in a few cases (Tiller and Buckley 1978). Previous patient series reported 2 patients with transient neutropenia and thrombocytopenia, 1 patient with persistent neutropenia (absolute neutrophils less than 0.5 × 109/L), and 1 patient who developed acute lymphoblastic leukemia (Dalal et al. 1998).
Developmental delay (17%) and congenital heart disease (13%) were also reported as THI manifestations in a series of 24 children with THI (Dorsey and Orange 2006).
Most of the reported patients with THI had normal or near normal antibody responses to immunization with tetanus and diphtheria toxoids, sometimes well before Ig levels became normal. They also had isohemagglutinin values within the normal range for age-matched controls, (Rieger et al. 1977; Tiller and Buckley 1978). Cano et al. (1990) demonstrated that 11 of 12 THI patients studied before the age of 17 months had made no specific antibodies to a panel of respiratory viruses despite recurrent upper respiratory infections. Resolution of the THI was marked by the appearance of specific viral antibodies, even before the serum IgG increased to normal levels (Cano et al. 1990). Unfortunately, data regarding specific antibodies to immunizations or isohemagglutinins are missing in that report.
In our series (Dalal et al. 1998), the ability to maintain protective levels of antibodies is as important as achieving the initial adequate response. Some of our patients showed an abnormal pattern in which they had a substantial but unsustained response to reimmunization despite normalizing total Ig concentrations. This type of dysgammaglobulinemia can be easily missed unless multiple determinations are obtained over a period of years.
Most studies have found that lymphocyte subpopulations and cellular immunity (as measured by proliferation assay in response to mitogens or specific antigens stimulation) are intact, including the level of memory and class-switched B cells (Rieger et al. 1977; Dressler et al. 1989; Kowalczyk et al. 1997; Dalal et al. 1998; Kilic et al. 2000). However, one study reported transient numeric and functional abnormalities of CD4 positive T helper cells (Moschese et al. 2007, 2008). Other investigators have found reduced frequencies of both circulating IgM+ and “switched” (IgM-IgD-) memory B cells and an inability to produce IgG in vitro in some children with THI; a phenotype that may identify the subset of patients whose immune defects persist (Moschese et al. 2007, 2008). This reduction in memory B cells has also been identified in children with early-onset common variable immunodeficiency (CVID) and selective IgA deficiency (Bukowska-Strakova et al. 2009) and older patients with CVID (Carsetti et al. 2005; Alachkar et al. 2006).
Differential diagnosis
By definition, THI is a self-limited disorder. Unfortunately, this diagnosis cannot be made with confidence until after a full laboratory and clinical recovery. Therefore, during this period, every effort should be made to differentiate THI from other primary IDs that could present in a similar manner such as X-linked agammaglobulinemia (XLA) or CVID. Typically, XLA can be easily differentiated from THI. The former is characterized by a severe deficiency of all Ig isotypes, an inability to produce antibodies, an absence of normal peripheral lymphoid tissues, an absence or a very low number of circulating B lymphocytes, and severe pyogenic infections starting in the first to second year of life (Buckley 1992).
Since the discovery that XLA is caused by mutations in the Bruton's tyrosine kinase gene (BTK) (Iseki and Heiner 1993), there have been increasing reports of patients who have atypical clinical and laboratory features that make the distinction between THI and XLA difficult to ascertain (Vetrie et al. 1993), underscoring the need for molecular screening of all patients with hypogammaglobulinemia for possible mutations in BTK (Kornfeld et al. 1995).
CVID, the most frequent type of hypogammaglobulinemia, is usually diagnosed in the second to third decade of life. Typically these patients are unable to produce protective levels of specific antibodies with variable immunoglobulin concentrations, with most having some impairment in cellular immunity (Buckley 1992, Iseki and Heiner 1993). In contrast, almost invariably, THI patients showed intact humoral and cellular immune function and normal antibody response.
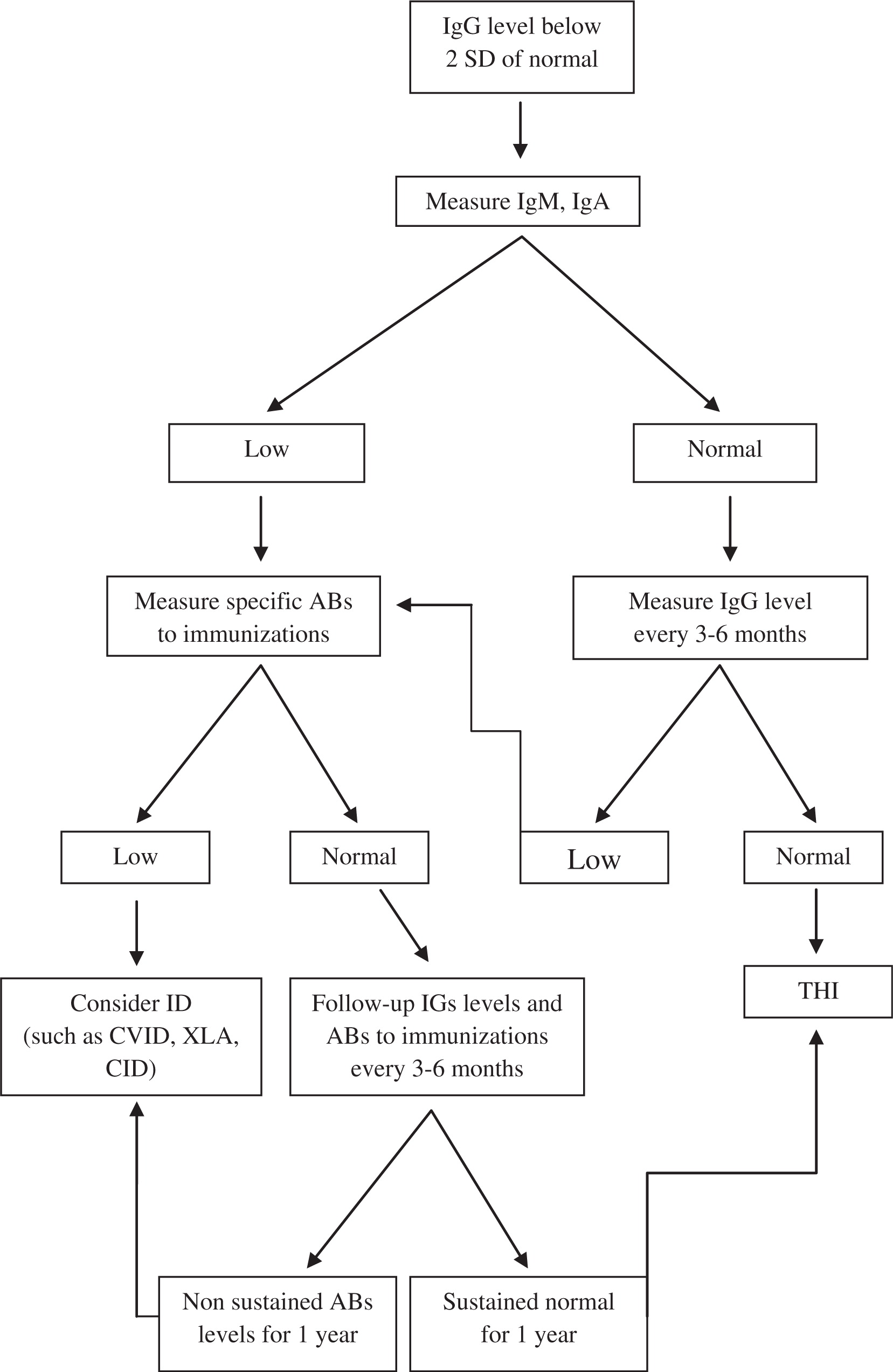
Our suggested algorithm for the diagnosis of THI is shown in Figure 1.
Figure 1:
Outcome
Although THI has been long recognized, little was known about the long-term outcome of these patients until our recent study (Dalal et al. 1998).
Previous studies predicted spontaneous clinical recovery by 9–15 months of age, and a rise to normal immunoglobulin levels by 2–4 years of age (Rosen and Janeway 1996; Wilson et al. 1996); however, by studying a limited number of patients, a few reports have shown that low IgA levels may persist beyond infancy up to the age of 5 years (Tiller and Buckley 1978; Benderly et al. 1986; Wilson et al. 1996).
In a prospective study, patients with hypogammaglobulinemia were followed from infancy. Specific antibody titers and clinical manifestations were recorded in 35 patients for 10 years. In all of these patients, the indication for immune evaluation was recurrent infections. The results clearly distinguish 3 different patterns of evolution (Figure 2). The first and most common pattern includes patients who experienced fewer infections as they grew older and eventually had normal total serum immunoglobulin levels, IgG subclass distribution, and specific antibody production. Some of these patients initially had low levels of specific antibodies but responded normally to reimmunization and were able to sustain these values. This process may span a decade and may include a transient phase whereby IgG subclasses may gradually normalize.
Figure 2:

The second group consisted of patients who continued to suffer repeated infections and whose IgG levels remained low and who were unable to mount significant antibody titers despite reimmunization. These patients subsequently required permanent intravenous immunoglobulin (IVIG) replacement therapy. Such patients can be classified as CVID in spite of the unusual presentation early in infancy.
The third pattern included patients who normalized their serum IgG levels but continued to experience significant infections. Upon reimmunization, they had a satisfactory but short-lived (declined within 1 year) response, despite having “normal” IgG levels. This group of patients therefore can be classified as dysgammaglobulinemia (Dalal et al. 1998).
Although it is impossible to predict at presentation the pattern patients will follow, invasive infections and low antibody levels at presentation appear to significantly predict ultimate permanent antibody deficiency (Dalal et al. 1998).
Recent studies have attempted to identify the prognostic markers that will predict the outcome of hypogammaglubulinemia in early childhood. Rutkowska et al. (2011, 2013) demonstrated that THI patients, unlike CVID patients, had increased levels of T regulatory cells and that may be used to differentiate between the 2 groups. Nonetheless, it is still not used as routine laboratory test.
In conclusion, THI is a diagnosis of exclusion that should be established in retrospect only after a long-term follow-up with thorough investigation of serial clinical and laboratory parameters.
Treatment
Symptomatic patients with repeated respiratory and/or ear infections may benefit from prophylactic antibiotic treatment. Rarely, in patients with recurrent severe infections who did not respond to antibiotic prophylaxis, temporary replacement therapy with IVIG is administered. (Cano et al. 1990; Wilson et al. 1996; Dalal et al. 1998).
REFERENCES
Adderson E.E., Shackelford P.G., Quinn A., and Carroll W.L. Restricted Ig H chain V gene usage in the human antibody response to Haemophilus influenza type b capsular polysaccharide J. Immunol. 1991 147 1667 -1674
Adenyi-Jones S.C., Faden H., Ferdon M.B., Kwong M.S., and Ogra P.L. Systemic and local immune responses to enhanced-potency inactivated poliovirus vaccine in premature and term infants J. Pediatr. 1992 120 686 -689
Agondi R.C., Barros M.T., Rizzo L.V., Kalil J., and Giavina-Bianchi P. Allergic asthma in patients with common variable immunodeficiency Allergy. 2010 65 510 -515
Alachkar H., Taubenheim N., Haeney M.R., Durandy A., and Arkwright P.D. Memory switched B cell percentage and not serum immunoglobulin concentration is associated with clinical complications in children and adults with specific antibody deficiency and common variable immunodeficiency Clin. Immunol. 2006 120 310 -318
Alford C.A., Stagno S., and Reynolds D.W. Diagnosis of chronic perinatal infections Am. J. Dis. Child. 1975 129 455 -463
Ballow M., Cates K.L., Rowe J.C., Goetz C., and Desbonnet C. Development of the immune system in very low birth weight (less than 1500 g) premature infants: Concentrations of plasma immunoglobulins and patterns of infection Pediatr. Res. 1986 20 899 -904
Benderly A., Pollack S., and Etzioni A. Transient hypogammaglobulinemia of infancy with severe bacterial infections and persistent IgA deficiency Isr. J. Med. Sci. 1986 22 393 -396
Bernbaum J.C., Daft A., Anolik R., Samuelson J., Barkin R., Douglas S., and Polin R. Response of preterm infants to diphtheria-tetanus-pertussis immunizations J. Pediatr. 1985 107 184 -188
Buckley R.H. Immunodeficiency diseases JAMA. 1992 268 2797 -2806
Bukowska-Straková K., Kowalczyk D., Baran J., Siedlar M., Kobylarz K., and Zembala M. The B-cell compartment in the peripheral blood of children with different types of primary humoral immunodeficiency Pediatr. Res. 2009 66 28 -34
Cano F., Mayo D.R., and Ballow M. Absent specific viral antibodies in patients with transient hypogammaglobulinemia of infancy J. Allergy. Clin. Immunol. 1990 85 510 -513
Carsetti R.I., Rosado M.M., Donnanno S., et al. The loss of IgM memory B cells correlates with clinical disease in common variable immunodeficiency J. Allergy. Clin. Immunol. 2005 115 412 -417
Dalal I., Reid B., Nisbet-Brown E., and Roifman C.M. The outcome of patients with hypogammaglobulinemia in infancy and early childhood J. Pediatr. 1998 133 144 -146
Dancis J., Osborn J.J., and Kunz H.W. Studies of immunology of the newborn infant, IV: Antibody formation in the premature infant Pediatrics. 1953 12 151 -157
Doğu F., Ikincioğullari A., and Babacan E. Transient hypogammaglobulinemia of infancy and early childhood: outcome of 30 cases Turk. J. Pediatr. 2004 46 120 -124
Dorsey M.J. and Orange J.S. Impaired specific antibody response and increased B-cell population in transient hypogammaglobulinemia of infancy Ann. Allergy. Asthma. Immunol. 2006 97 590 -595
Dressler F., Peter H.H., Müller W., and Rieger C.H. Transient hypogammaglobulinemia of infancy: Five new cases, review of the literature and redefinition Acta. Paediatr. Scand. 1989 78 767 -774
Einhorn M.S., Granoff D.M., Nahm M.H., Quinn A., and Shackelford P.G. Concentrations of antibodies in paired maternal and infant sera: Relationships to IgG subclass J. Pediatr. 1987 111 783 -788
Fudenberg H.H. and Fudenberg B.R. Antibody to hereditary human gamma globulin (Gm) factor resulting from maternal-fetal incompatibility Science. 1964 145 170 -171
Gitlin D. and Janeway C.A. Agammaglobulinemia: Congenital, acquired and transient forms Prog. Hematol. 1956 1 318 -329
Hayakawa H., Iwata T., Yata J., and Kobayashi N. Primary immunodeficiency syndrome in Japan. I. Overview of a nationwide survey on primary immunodeficiency syndrome J. Clin. Immunol. 1981 1 31 -39
Hobbs J.R. and Davis J.A. Serum G-globulin levels and gestational age in premature babies Lancet. 1967 1 757 -759
Iseki M. and Heiner D.C. Immunodeficiency disorders Pediatr. Rev. 1993 14 226 -236
Karaca N.E., Aksu G., Gulez N., Yildiz B., Azarsiz E., and Kutukculer N. New laboratory findings in Turkish patients with transient hypogammaglobulinemia of infancy Iran. J. Allergy. Asthma. Immunol. 2010 9 237 -243
Keles S., Artac H., Kara R., Gokturk B., Ozen A., and Reisli Transient hypogammaglobulinemia and unclassified hypogammaglobulinemia: ‘similarities and differences’ Pediatr. Allergy. Immunol. 2010 21 843 -851
Kidon M.I., Handzel Z.T., Schwartz R., Altboum I., Stein M., and Zan-Bar I. Symptomatic hypogammaglobulinemia in infancy and childhood - clinical outcome and in vitro immune responses BMC Fam. Pract. 2004 2 5 23
Kiliç S.S., Tezcan I., Sanal O., Metin A., and Ersoy F. Transient hypogammaglobulinemia of infancy: clinical and immunologic features of 40 new cases Pediatr. Int. 2000 42 647 -650
Koblin B.A., Townsend T.R., Muñoz A., Onorato I., Wilson M., and Polk B.F. Response of preterm infants to diphtheria-tetanus pertussis vaccine Pediatr. Infect. Dis. J. 1988 7 704 -711
Kohler P.F. and Farr R.S. Elevation of cord over maternal IgG immunoglobulin: Evidence for an active placental IgG transport Nature. 1966 210 1070 -1071
Kornfeld S.J., Kratz J., Haire R.N., Litman G.W., and Good R.A. X-linked agammaglobulinemia presenting as transient hypogammaglobulinemia of infancy J. Allergy. Clin. Immunol. 1995 95 915 -917
Kowalczyk D., Mytar B., and Zembala M. Cytokine production in transient hypogammaglobulinemia and isolated IgA deficiency J. Allergy. Clin. Immunol. 1997 100 556 -562
McGeady S.J. T ransient hypogammaglobulinemia of infancy: Need to reconsider name and definition J. Pediatr. 1987 110 47 -50
McNabb T., Koh T.Y., Dorrington K.J., and Painter R.H. Structure and function of immunoglobulin domains. V. Binding of immunoglobulin G and fragments to placental membrane preparations J. Immunol. 1976 117 882 -888
Moschese V., Carsetti R., Graziani S., et al. Italian primary immunodeficiency network: Memory B-cell subsets as a predictive marker of outcome in hypogammaglobulinemia during infancy J. Allergy. Clin. Immunol. 2007 120 474 -476
Moschese V., Graziani S., Avanzini M.A., et al. A prospective study on children with initial diagnosis of transient hypogammaglobulinemia of infancy: results from the Italian Primary Immunodeficiency Network Int. J. Immunopathol. Pharmacol. 2008 21 343 -352
Nathenson G., Schorr J.B., and Litwin S.D. Gm factor fetomaternal gamma globulin incompatibility Pediatr. Res. 1971 5 2 -9
Ochs H.D. and Wedgewood R.J. IgG subclass deficiencies Annu. Rev. Med. 1987 38 325 -340
Papadopoulou A., Mermiri D., Taousani S., Triga M., Nicolaidou P., and Priftis K.N. Bronchial hyper-responsiveness in selective IgA deficiency Pediatr. Allergy. Immunol. 2005 16 495 -500
Qian J.H., Zhu J.X., Zhu X.D., and Chen T.X. Clinical features and follow-up of Chinese patients with symptomatic hypogammaglobulinemia in infancy Chin. Med. J. 2009 122 1877
Report of an IUIS Scientific Committee: Primary immunodeficiency diseases Clin. Exp. Immunol. 1999 118 Suppl 1 1 -28
Rieger C.H., Nelson L.A., Peri B.A., Lustig J.V., and Newcomb R.W. Transient hypogammaglobulinemia of infancy J. Pediatr. 1977 91 601 -603
Rosen F.S. and Janeway C.A. The gammaglobulins: III. The antibody deficiency syndromes N. Engl. J. Med. 1966 275 769 -775
Rutkowska M., Lenart M., Bukowska-Strakovà K., et al. The number of circulating CD4+ CD25high Foxp3+ T lymphocytes is transiently elevated in the early childhood of transient hypogammaglobulinemia of infancy patients Clin. Immunol. 2011 140 307 -310
Rutkowska M., Trzyna E., Lenart M., et al. The elevated number of circulating regulatory T cells in patients with transient hypogammaglobulinemia of infancy is not associated with any abnormalities in the genes encoding the TGF-β receptors Clin. Immunol. 2013 149 83 -85
Shapiro R., Beatty D.W., Woods D.L., and Malan A.F. Serum complement and immunoglobulin values in small-for-gestational-age infants J. Pediatr. 1981 99 139 -141
Siegel R.L., Issekutz T., Schwaber J., Rosen F.S., and Geha R.S. Deficiency of T helper cells in transient hypogammaglobulinemia of infancy N. Engl J. Med. 1981 305 1307 -1313
Smith D.H., Peter G., Ingram D.L., Harding A.L., and Anderson P. Responses of children immunized with the capsular polysaccharide of Haemophilus influenzae Pediatrics. 1973 52 637 -644
Smolen P., Bland R., Heiligenstein E., Lawless M.R., Dillard R., and Abramson J. Antibody response to oral polio vaccine in premature infants J. Pediatr. 1983 103 917 -919
Soothill J.F. Immunoglobulins in first-degree relatives of patients with hypogammaglobulinemia Lancet. 1968 1 1001 -1003
Tiller T.L. and Buckley R.H. Transient hypogammaglobulinemia of infancy: Review of the literature, clinical and immunologic features of 11 new cases, and long-term follow-up J. Pediatr. 1978 92 347 -353
Van de Winkel J.G.J. and Anderson C.L. Biology of human immunoglobulin G Fc receptors J. Leukoc. Biol. 1991 49 511 -524
Van Winkle R.C.L., Hauck W.W., and McGeady S.J. Phenotypic parameters predict time to normalization in infants with hypogammaglobulinemia J. Clin. Immunol. 2013 33 1336 -1340
Vetrie D., Vořechovský I., Sideras P., et al. The gene involved in X-linked agammaglobulinemia is a member of the src family of proteintyrosine kinases Nature. 1993 361 226 -233
Walker A.M., Kemp A.S., Hill D.J., and Shelton M.J. Features of transient hypogammaglobulinemia in infants screened for immunological abnormalities Arch. Dis. Child. 1994 70 183 -186
Whelan M.A., Hwan W.H., Beausoleil J., Hauck W.W., and McGeady S.J. Infants presenting with recurrent infections and low immunoglobulins: characteristics and analysis of normalization J. Clin. Immunol. 2006 26 7 -11
Wilson, C.B., Lewis, D.B., and Penix, L.A. 1996. The physiologic immunodeficiency of immaturity. In Immunologic Disorders in Infants and Children. Edited by E.R. Stiehm. Philadelphia, WB Saunders, p 253.
World Health Organization Primary immunodeficiency diseases: Report of a WHO scientific group Immunodefic. Rev. 1992 3 195 -236
Information & Authors
Information
Published In

LymphoSign Journal
Volume 01 • Number 01 • September 2014
Pages: 1 - 9
History
Received: 1 July 2014
Accepted: 2 July 2014
Version of record online: 20 August 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
OvadiaAdi and DalalIlan. 2014. Transient hypogammaglobulinemia of infancy. LymphoSign Journal.
01(01): 1-9. https://doi.org/10.14785/lpsn-2014-0006
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Get Access
Login options
Check if you access through your login credentials or your institution to get full access on this article.
